はじめに
RNA-seq解析では、解析の過程で様々な種類のファイルと解析ツールが登場するので、「それぞれのファイルには何の情報が格納されているのか?」、「どの解析ツールを使えばいいのか?」「次の解析は何をしたらいいのか?」など、初学者がつまずきやすい箇所がたくさんあります。
そこで今回はドライ解析入門者の方を対象に、RNA-seq解析(ドライ解析)の基本的な手順について解説していきます。特に、ヒトやマウスなどの検体を用いて発現変動遺伝子解析を行うことを想定して、RNA-seq解析のゴールドスタンダードを詳しく解説していきます!ぜひ最後までご覧ください!
実際には実験の目的に合わせて解析方法を変える必要があるのですが、本記事では最も広く使用されている標準的な方法を紹介していきます!
Illuminaシークエンサーを含む次世代シークエンサーのシークエンス原理については下記の記事で詳しく解説しています。合わせてご覧ください。
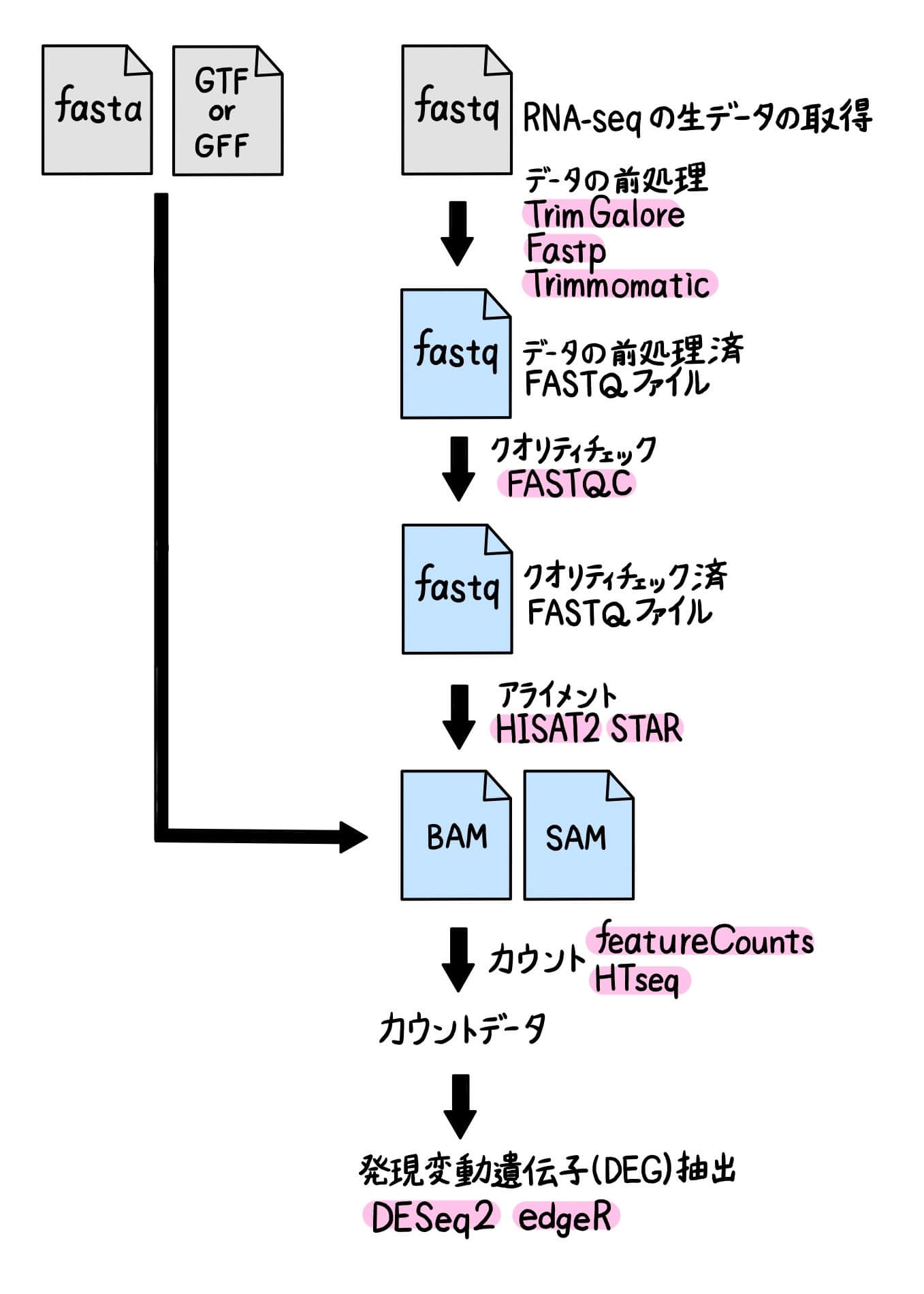
RNA-seqのワークフロー(ドライ解析)
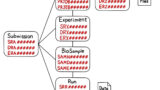
RNA-seq解析全体の流れを図1に示しました。次項から詳しく解説していきますので、まずは図1で全体像を把握しましょう。また、RNA-seq解析の過程で使用されるファイル形式を表1にまとめております。
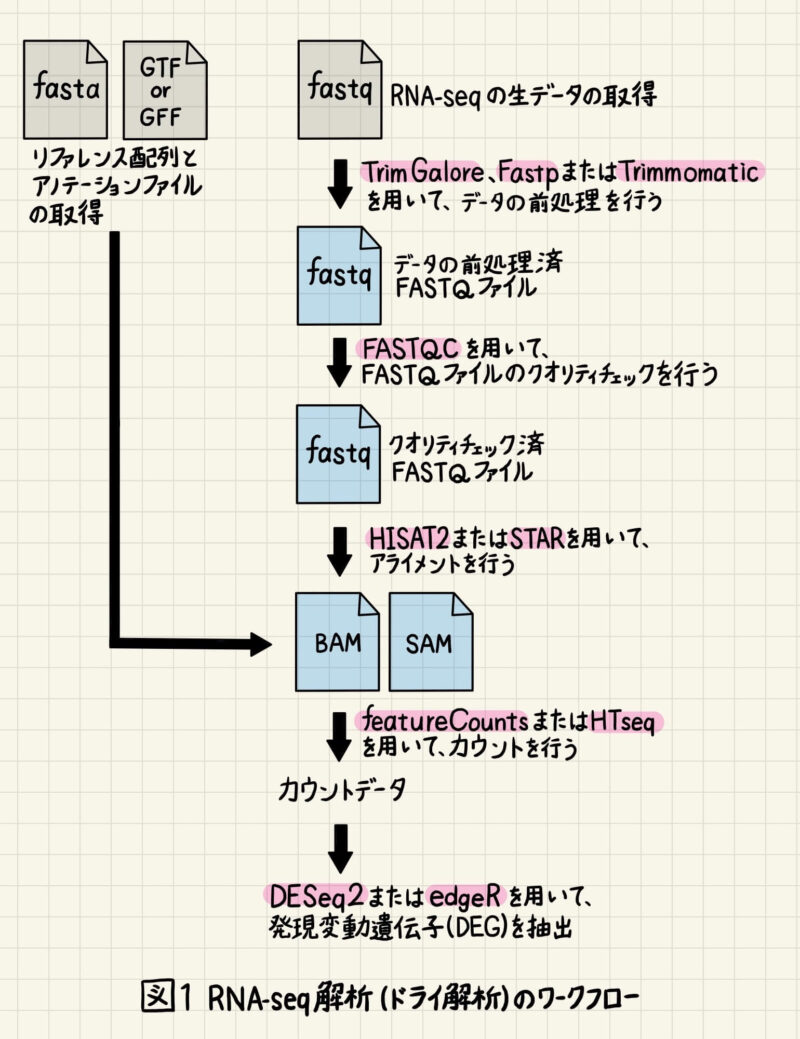
表1 RNA-seq解析で使用される代表的なファイル形式
| ファイル名 | 内容 |
| FASTQ | 次世代シークエンサーから出力される塩基配列データはFASTQ形式で出力される。FASTQファイルは4行が1セットになっており、1行目に「@sequence_ID(先頭は@で始まる)」、二行目に塩基配列、三行目に「+(プラス)記号」、四行目に「クオリティスコア」が含まれる。 |
| FASTA | FASTAファイルも塩基配列が格納されているが、「クオリティスコア」が含まれていない。リファレンス配列はFASTA形式で保存されている。 |
| GTF/GFF | 遺伝子の位置や機能などのアノテーション情報が格納されているファイル。 |
| BAM | リードがゲノム上のどの位置にマッピングされたかを格納しているバイナリ形式のファイル。 |
| SAM | BAMファイルを人が読めるようにテキスト形式に変換したファイルをSAMファイルという。 |
fastqファイルの取得
次世代シークエンサーから得られるシークエンスデータはfastq形式で出力されます。fastqファイルの拡張子は「.fastq」または「.fq」で、しばしばgzip圧縮されており、「.fastq.gz」または「.fq.gz」となっている場合があります。fastqファイルは4行が1セットになっており、1行目に「@リード名(先頭は@で始まる)」、二行目に塩基配列、三行目に「+(プラス)記号」、四行目に「クオリティスコア」が含まれています(図2)。
@read name
AGCTGTACGTATCGACTGAGTCGTAG
+
7>I:B;I?8$I>?9I;?<I;5E>?;;図2 FASTQファイルの中身の例
公共のデータベースに登録されているデータを使用する場合は、SRA Toolkitというツールに含まれるprefetchを用いてSRA形式のファイルをダウンロードします。そしてfasterq-dump(fastq-dumpの後継版)を用いてSRAファイルをfastqファイルに変換します。
fasterq-dumpを使って、fastqファイルを直接ダウンロードすることも可能です。prefetchを用いた方法を紹介したのは、ダウンロード速度などの点で優れているからです。ダウンロードにprefetchを用いることでSRAファイルを効率的にダウンロードできます。
SRAのデータ構造など、SRAに関する基本的な知識は下記の記事で詳しく解説しています。
データの前処理
次世代シークエンサーから出力されたfastqファイルは、アダプター配列や低品質なリードを含んでいる可能性があります。そのため、Trim Galore、Fastp、Trimmomaticなどのツールを用いて、不要な配列の除去やクオリティフィルタリングなどの前処理を行う必要があります。 Trim Galore、Fastp、Trimmomaticはいずれも広く使用されており、基本的にどのツールを用いても良いです。
前処理が終了したfastqファイルは、元のファイルと同じfastq形式で出力されます。
前処理済fastqファイルのクオリティチェック
データの前処理が終了した後は、FASTQCを用いて前処理済fastqファイルのクオリティチェックを行います。FASTQCは、fastq形式のシークエンスデータの品質を11の項目で評価し、それぞれの項目に“pass”、“warning”、“fail”のいずれかの評価を付けます。評価結果はHML形式で出力されます。
確認すべき評価項目は実験の目的によって異なるため、特定の項目で“fail”評価受けていても、必ずしもシークエンスのやり直しを意味している訳ではないことに留意が必要です。
アラインメント
リードの配列と既知の配列(リファレンス配列)を比較し、リードがどこのDNA領域に由来するかを特定するプロセスをアライメントまたはマッピングと言います。リファレンス配列にはゲノムまたはトランスクリプトームが使用されますが、通常のRNA-seq解析ではリファレンスゲノムに対してアラインメントを行います。ヒトやマウスのように、リファレンストランスクリプトームが入手可能な場合は、リファレンス配列にトランスクリプトームを用いる場合もあります。
インデックスファイルの作成
アライメントステップでは、リードをリファレンス配列にマッピングする前に、インデックスファイルを作成する必要があります。インデックスファイルは、リファレンス配列から配列データを効率的に検索し、 アライメント位置を特定するために必要なファイルです。インデックスファイルを作成した後、後述のアライメントツールを用いてマッピングを行なっていきます。
アライメントに使用するインデックスファイルは、リファレンス配列及びアノテーション情報から作成します。リファレンス配列及びアノテーション情報は、GENCODE、Ensembl、RefSeq、UCSC Genome Browserのいずれかのデータベースから入手します。RNA-seq解析の場合は、一般的にGENCODEまたはEnsemblを使用します。GENCODEとEnsemblはほぼ同じアノテーション情報を提供しており、どちらを使用しても構いません。
アノテーション情報には、遺伝子の位置や機能などの情報が含まれており、正確なアライメント位置の特定に必要です。
リファレンス配列はfasta形式で提供されます。fastaファイルは、配列の品質情報を含んでいない点でfastqファイルと異なっています。また、アノテーションファイルは「.gtf」や「.gff」で提供されています。
リファレンス配列へのアライメント
RNA-seqではイントロンが除去された成熟mRNAの塩基配列を読んでいるため、一つのリードが複数のエキソンにまたがっている場合があります。したがって、リードをゲノムにアラインメントする場合は、RNAスプライシングを考慮したアライメントツール(splice-aware aligner)を使用する必要があります。現在数多くのsplice-aware alignerがありますが、RNA-seq解析で頻繁に使用されるのはHISAT2またはSTARです(表2)。その他のアライメントツールとして、BWAやBowtie2も有名ですが、これらのツールはもともとDNAリードをゲノムにマッピングする目的で開発されたものであり、通常はRNA-seq解析に特化したツール(HISAT2またはSTAR)を使用した方が良いでしょう。
表2 RNA-seq解析で使用される代表的なアライメントツール
| ツール名 | 特徴 |
| HISAT2 | ・splice-aware alignerのアライメントツール ・TopHat2及びHISATの後継版。 ・STARよりもメモリ消費量が少ない。 |
| STAR | ・splice-aware alignerのアライメントツール。 ・大量のメモリを消費するが、HISAT2と比較してマッピング率、アライメント速度、正確性に優れる。 |
HISAT2やSTARなどアライメントツールは、アライメント結果をSAMまたはBAM形式で出力します。SAMは人が読めるようなテキスト形式のファイルで、BAMはSAMファイルをバイナリ形式に変換したファイルです。SAMファイルよりも、BAMファイルの方がファイルサイズが小さいです。
最近はリファレンス配列にアラインメントを行わないアライメントフリーの解析ツール(Salmon及びkallisto)も開発されています(表3)。Salmonとkallistoは類似の解析ツールで、従来のアライメントベースの解析ツールと比較してはるかに高速な点が特徴です。 Salmonとkallistoが行う近似的なアライメントは、それぞれpseudo-alignment(擬似アラインメント) 、quasi-mappingと呼ばれます。 また、アライメントを行わないので、SAMファイルやBAMファイルを生成しません。すなわち、Salmon及びkallistoはfastqファイルから直接カウントテーブルを出力します。
アライメント情報はアライメントのクオリティチェックや、アライメント情報を用いた詳細な解析に必要な場合があり、研究の目的によって使い分ける必要があります。
表3 アライメントフリーの発現量解析ツール
| ツール名 | 特徴 |
Salmon、kallisto | ・トランスクリプトームをリファレンスとして使用する。 ・アライメントフリーで発現量の定量を行うので、非常に高速。 ・k-mer[1]ベースのカウントを行い、Expectation Maximization (EM) アルゴリズムを用いて発現量を推定する。 ・新規アイソフォームの検出はできない。 |
[1] k-mer(ケーマーと発音する); リード中のk個の連続した塩基のこと。 Salmon及びkallistoは各k-merとトランスクリプトームとの一致度を評価する。
カウント
続いて、リファレンス配列にアラインメントされたリードをカウントツールを用いてカウントしていきます。カウントツールは数多くありますが、その中でも特によく使用されているのがfeatureCountsとHTSeqです(表4)。
表4 RNA-seq解析で使用される代表的なカウントツール
| ツール名 | 特徴 |
| featureCounts、HTSeq | DNAの領域には、遺伝子やエキソン、プロモーターなどの特徴(feature)がアノテーション情報として付けられている。featureCounts及びHTSeqでは、アノテーションが付けられた特定の領域とリードが重なっていれば、カウントを行う。 |
発現変動遺伝子(DEG)の抽出
featureCountsやHTSeqを用いてリード数をカウントした後は、サンプル間または遺伝子間で比較ができるようにデータを正規化し、発現変動遺伝子(differentially expressed gene; DEG)の検出を行います。データの正規化・発現変動遺伝子の検出に使用される代表的なツールはDESeq2とedgeRです(表5)。ESeq2及びedgeRはRのパッケージで、featureCountsやHTSeqが出力したカウントデータ(テキスト形式のファイル)を読み込み、DEGの検出を行います。
DEGの抽出完了後は、Rを用いてMA plot、Volcano plot、ヒートマップ等でDEGを可視化したり、GO解析やPathway解析などを実行したり、実験の目的に合わせた解析を行っていきます。
| ツール名 | 特徴 |
| DESeq2 | 正規化方法:DESeq’s size factor |
| edgeR | 正規化方法:Trimmed Mean of M-values (TMM) |
リードカウントは総リード数と遺伝子長に依存します。つまり、総リード数が多いサンプル及び遺伝子長が長い遺伝子ほど、マッピングされるリードが多くなってしまいます。このままでは、サンプル間、遺伝子間で発現量の比較をすることができないので、データを正規化する必要があります。
異なるサンプル間での比較をするには、DESeq’s size factor、Trimmed Mean of M-values (TMM) などの手法を用いてデータを正規化します。同じサンプル内で異なる遺伝子発現量の比較をするには、RPKM (Reads Per Kilobase of exon per Million mapped reads) 、FPKM (Fragments Per Kilobase of exon per Million mapped reads) 、TPM(transcripts per million)などの手法を使って正規化をします。 ただ、RPKM及びFPKMは適切に遺伝子発現量を表現していないことが指摘されており、最近はRPKM、FPKMよりもTPMの使用が推奨されています。
以上をまとめると、異なるサンプル間での比較においては、バッチ効果などが考慮されたDESeq’s size factorまたはTMMを使用し、同じサンプル内の比較であればTPMを用いるのが一般的です。
まとめ
- 次世代シークエンサーから出力されるFASTQファイルを入手する。
- Trim Galore、Fastp、Trimmomaticなどを用いて、FASTQファイルに含まれる不要な配列や質の低いデータを除く。
- FASTQCを用いて、FASTQファイルのクオリティチェックを行う。
- リファレンス配列(FASTAファイル)とアノテーションファイル(GTF/GFF)から、インデックスファイルを作成する。
- HISAT2またはSTARを用いて、クオリティチェック済のFASTQファイルをリファレンス配列にマッピングする。
- featureCountsまたはHTSeqを用いて、マッピングデータ(BAMファイル)からマッピングされたリード数をカウントする。(※Salmon及びkallistoはアライメントを行わず、FASTQから直接カウントデータを出力する。)
- DESeq2またはedgeRを用いて、カウントデータから発現変動遺伝子(DEG)を抽出する。
- Zhao S, Zhang Y, Gamini R, Zhang B, von Schack D. Evaluation of two main RNA-seq approaches for gene quantification in clinical RNA sequencing: polyA+ selection versus rRNA depletion. Sci Rep. 2018 Mar 19;8(1):4781. doi: 10.1038/s41598-018-23226-4. PMID: 29556074; PMCID: PMC5859127.
- Zhao, S., Zhang, Y., Gordon, W., Quan, J., Xi, H., Du, S., von Schack, D., & Zhang, B. (2015). Comparison of stranded and non-stranded RNA-seq transcriptome profiling and investigation of gene overlap. BMC genomics, 16(1), 675. https://doi.org/10.1186/s12864-015-1876-7
- Raplee ID, Evsikov AV, Marín de Evsikova C. Aligning the Aligners: Comparison of RNA Sequencing Data Alignment and Gene Expression Quantification Tools for Clinical Breast Cancer Research. J Pers Med. 2019 Apr 3;9(2):18. doi: 10.3390/jpm9020018. PMID: 30987214; PMCID: PMC6617288.
- Musich, Ryan J. A Recent (2020) Comparative Analysis of Genome Aligners Shows HISAT2 and BWA are Among the Best Tools. Rochester Institute of Technology, 2020.