はじめに
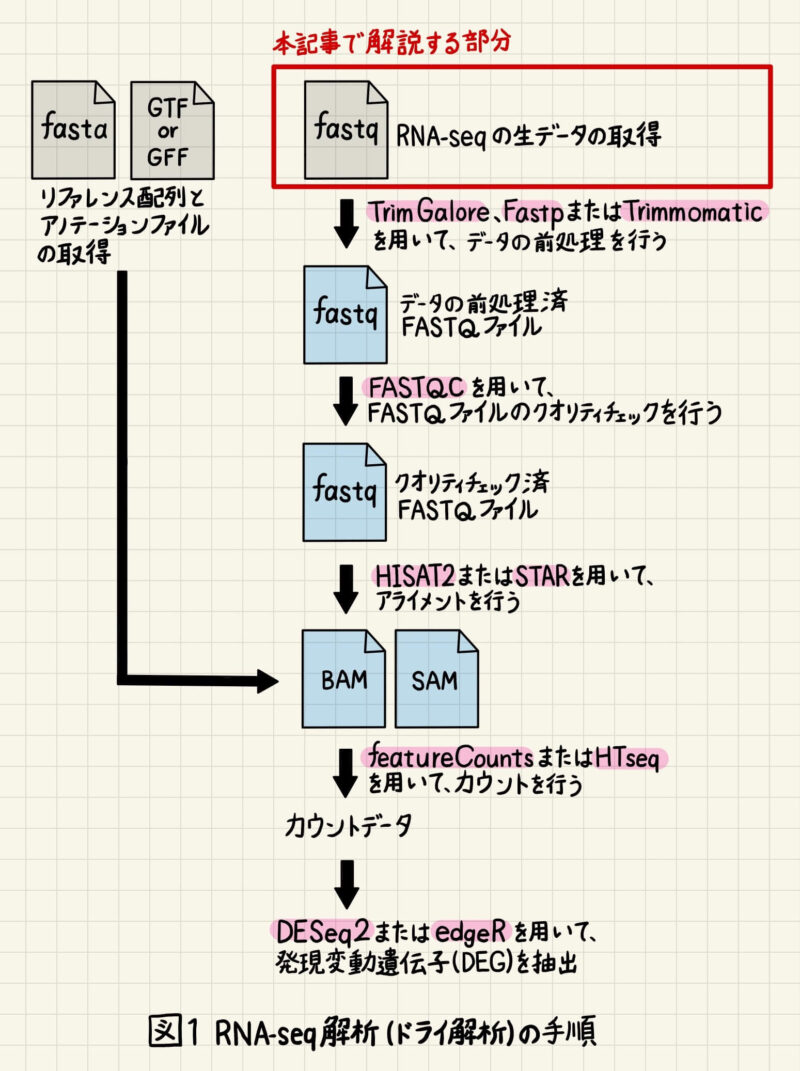
今回は公共データベースに保存されているsra形式のシークエンスデータをダウンロードし、fastq形式のファイルを取得する方法について、実際の論文データを用いて詳しく解説していきます(図1)。誰でも再現できるように使用するコマンドについても丁寧に解説していきますので、ぜひ最後までご覧ください!
著者の実行環境は下記の通りです。
- PC: MacBook Pro 14インチ、2021
- チップ: Apple M1 Pro
- メモリ: 32 GB
- OS: macOS Ventura 13.4.1
- ストレージ: 512 GB
- ターミナル: zsh
※RNA-seq解析(ドライ解析)全体の流れは下記の記事で詳しく解説しています。合わせてご覧ください。
SRAからFASTQをダウンロードする方法の概要
次世代シークエンサーから出力されるシークエンスデータはfastqという形式で、RNA-seq解析にはFASTQファイルを必要とします。FASTQファイルはsra形式で公共データベースに保存されていますので、公共データベースを使用する場合はまずSRAファイルを入手し、その後FASTQファイルに変換する必要があります(図2)。本記事ではこのFASTQファイルを取得するまでの手順を、実際の論文データを用いて詳しく解説していきます。

シークエンスデータは下記の論文で実施されたRNA-seqのデータを使用します。この論文では、「ヒト胎児横紋筋肉腫(rhabdomyosarcoma; RD)細胞」及び「EHMT2という遺伝子をsiRNAによりKD(ノックダウン)したRD細胞」の2群(各n = 3、計6サンプル)で、RNA-seq解析を行っています。
使用するデータ
Pal, A., Leung, J. Y., Ang, G. C. K., Rao, V. K., Pignata, L., Lim, H. J., Hebrard, M., Chang, K. T., Lee, V. K., Guccione, E., & Taneja, R. (2020). EHMT2 epigenetically suppresses Wnt signaling and is a potential target in embryonal rhabdomyosarcoma. eLife, 9, e57683. https://doi.org/10.7554/eLife.57683
SRAファイルの探し方
次世代シークエンシングの解析結果を論文に投稿する際は、シークエンスデータををNCBI(National Center for Biotechnology Information)、DDBJ(DNA Data Bank of Japan)、EBI(European Bioinformatics Institute)のいずれかのデータベースに登録することが義務付けられています。公共データベースには、シークエンスデータ(SRAファイル)だけでなく、実験目的、使用した機器、解析結果などのデータも格納されています。これらのデータにはそれぞれ異なるアクセッション番号が割り振られており、初めて見るとどこに何のデータが格納されているのか非常に分かりづらいです。ですので、まずは論文中のアクセッション番号からシークエンスデータを探していくところから解説していきます。
※公共データベース(NCBI、DDBJ、EBI)から自分の興味のあるシークエンスデータを入手するためには、SRAファイルのデータ構造を理解していないと難しいです。下記の記事でSRAファイルのデータ構造を詳しく解説していますので、合わせてご覧ください。
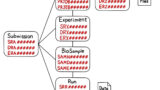
論文中の「Data availability」項を見ると、この論文で実施されたRNA-seqのシークエンスデータは「GSE142975」というアクセッション番号でGEO(Gene Expression Omnibus)
に保存されていることがわかります。GEOとは、NCBIが管理・運用しているNGS(next-generation sequencing)データベースのことです。GEOのサイトを見るといろいろなアクセッション番号が出てきますが、シークエンスデータが格納されているファイルは「SRR########」というファイルです。「SRR########」がどこにあるかというと、今回の論文では、 「GSM4247414」→「GSE142975」→「SRX7495744」→「SRR10822355」のようにアクセッション番号を辿っていくとSRRから始まるデータを見つけることができます。これは6つあるシークエンスデータの一つですが、残りのシークエンスデータも同様に探していきます(表1)。
ちなみに、「GSE#######」にはサンプル情報、遺伝子発現データ、メタデータなどが格納されています。また、「SRX#######」には解析に用いた次世代シークエンサーの機器名、ライブラリの調製方法、リード数などのRUN情報が格納されています。
表1 解析で使用するサンプルのアクセッション番号
| 群 | サンプル名 | GSE | SRX | SRR |
| Ctrl | RD cells, G9acontrol1 | GSM4247414 | SRX7495744 | SRR10822355 |
| Ctrl | RD cells, G9acontrol2 | GSM4247415 | SRX7495745 | SRR10822356 |
| Ctrl | RD cells, G9acontrol3 | GSM4247416 | SRX7495746 | SRR10822357 |
| KD | RD cells, KnockDown_G9a1 | GSM4247417 | SRX7495747 | SRR10822358 |
| KD | RD cells, KnockDown_G9a2 | GSM4247418 | SRX7495748 | SRR10822359 |
| KD | RD cells, KnockDown_G9a3 | GSM4247419 | SRX7495749 | SRR10822360 |
FASTQファイルの取得方法
シークエンスデータのアクセッション番号がわかりましたので、SRAファイルをダウンロードしていきましょう。
Anacondaのダウンロード
まずは、Anacondaを下記のAnaconda公式ページからダウンロードします。 Anacondaはデータサイエンスや機械学習などで頻繁に使用されるパッケージやツールを多く組み込んだ ”機能強化版” Pythonです。このように機能が追加されたPythonのことをPythonディストリビューションと言い、AnacondaはPythonディストリビューションの一つです。
Anaconda公式ページ: https://www.anaconda.com/
Anacondaをダウンロードすることで、condaという拡張機能が利用できるようになります。condaはAnacondaに含まれているツールの一つで、本家版Pythonのpipコマンドではインストールすることができないライブラリを簡単にインストールすることができます。
このcondaコマンドを用いて、SRA Toolkitをダウンロードします。SRA Toolkitをダウンロードすることで、SRA形式のファイルをダウンロードするprefetchや、SRAファイルをfastqファイルに変換するfasterq-dump(fastq-dumpの後継版)などのツールを使用することができるようになります。
# Anacondaに含まれるcondaコマンドで、SRA Toolkitをダウンロード
% conda install -c bioconda sra-tools※ %は行頭を表すプロンプト記号なので、入力しないでください。
condaコマンドでエラーが出る場合
2020年よりMacBookではM1、M2チップが採用されています。その影響で、condaコマンドを用いてSRA Toolkitをインストールしようとしても、パッケージが見つからないというエラーが出るようです。
実際に私の実行環境で、SRA Toolkitをインストールしようとすると下記のようなエラーメッセージが出ます。
% conda install -c bioconda sra-tools
Collecting package metadata (current_repodata.json): done
Solving environment: unsuccessful initial attempt using frozen solve. Retrying with flexible solve.
Collecting package metadata (repodata.json): done
Solving environment: unsuccessful initial attempt using frozen solve. Retrying with flexible solve.
PackagesNotFoundError: The following packages are not available from current channels:
- sra-tools
Current channels:
- https://conda.anaconda.org/bioconda/osx-arm64
- https://conda.anaconda.org/bioconda/noarch
- https://repo.anaconda.com/pkgs/main/osx-arm64
- https://repo.anaconda.com/pkgs/main/noarch
- https://repo.anaconda.com/pkgs/r/osx-arm64
- https://repo.anaconda.com/pkgs/r/noarch
To search for alternate channels that may provide the conda package you're
looking for, navigate to
https://anaconda.org
and use the search bar at the top of the page.このようなエラーが出る場合は、macOS向けのパッケージ管理ツールであるHomebrewを代わりに使用しましょう。Homebrewでもcondaと同様、バイオインフォマティクスのツールをインストールすることができます。
Homebrewのダウンロード方法、Homebrewを用いたSRA Toolkitのインストール方法は下記の通りです。
# Homebrewのインストール
% /bin/bash -c "$(curl -fsSL https://raw.githubusercontent.com/Homebrew/install/HEAD/install.sh)”
# brewコマンドでsratoolkitをインストール
% brew install sratoolkitSRAファイルの取得
SRAファイルのダウンロードに必要なツールをインストールすることができましたので、続いてSRAファイルのダウンロード先のディレクトリを作成しておきます。
Macのターミナルを開くと、 下記のように表示されます。usernameという名前のユーザが、MacBook-Proを使用しているという意味です。
username@MacBook-Pro ~ %「~(チルダ)」は、「ホームディレクトリ」を意味しています。ホームディレクトリとはユーザがログインした直後のディレクトリを意味しています。ディレクトリはフォルダと同じ意味ですが、プログラミング関連の話ではフォルダをディレクトリと呼称する場合が多いです。
それではホームディレクトリの直下にSRAを保存するディレクトリを作ります。今回は「GSE142975」というアクセッション番号のデータですので、「GSE142975」という名前のディレクトリを作成し、そこにシークエンスデータを入れていくことにします。
# 「GSE142975」というディレクトリ作成
% mkdir GSE142975
# 「GSE142975」というディレクトリ移動
% cd GSE142975
# 現在のパスを表示して、カレントリディレクトリを確認
% pwd
/Users/username/GSE142975続いて、touchコマンドでテキストファイルを作成し、そこにダウンロードしたいSRR番号を改行区切りで記述します。
そして、SRA Toolkitに含まれるprefetchコマンドでテキストファイルに記述されているSRRをまとめてダウンロードします。
# 「GSE142975.txt」という名前のテキストファイルを作成
% touch GSE142975.txt
# openコマンドで「GSE142975.txt」を開き、改行区切りで「SRR10822355」、「SRR10822356」、「SRR10822357」、「 SRR10822358」、「 SRR10822359」「SRR10822360」を記述
% open GSE142975.txt
# catコマンドで「GSE142975.txt」の中身を確認
% cat GSE142975.txt
SRR10822355
SRR10822356
SRR10822357
SRR10822358
SRR10822359
SRR10822360
# SRA Toolkitに含まれるprefetchコマンドで、「GSE142975.txt」に記述したSRRをまとめてダウンロード
% prefetch --option-file GSE142975.txtfastqファイルの取得方法
取得したSRAファイルを、fasterq-dumpコマンドを用いてFASTQに変換します。
# SRAファイルをFASTQファイルに変換
% fasterq-dump SRR10822355 SRR10822356 SRR10822357 SRR10822358 SRR10822359 SRR10822360
# ディレクトリの中身を確認
% ls
GSE142975.txt SRR10822355_1.fastq SRR10822355_2.fastq SRR10822356_1.fastq SRR10822356_2.fastq SRR10822357_1.fastq SRR10822357_2.fastq SRR10822358_1.fastq SRR10822358_2.fastq SRR10822359_1.fastq SRR10822359_2.fastq SRR10822360_1.fastq SRR10822360_2.fastq1つのSRAファイルから「SRR########_1.fastq」と「SRR########_2.fastq」の2つのFASTQファイルが生成されたことがわかります。これは、このRNA-seqはペアエンドで行われているということを意味しています。
FASTQファイルの圧縮
FASTQファイルはサイズが大きいので、pigzコマンドを用いてgzip圧縮します。今回のFASTQファイルは14GB程度ありましたが、gzip圧縮により約3GBにまで圧縮できました。
# pigzをインストール
% conda install pigz
# カレントディレクトリにあるFASTQファイルを全てgzip圧縮
% pigz *fastq
# ディレクトリの中身を確認
% ls
GSE142975.txt SRR10822355 SRR10822355_1.fastq.gz SRR10822355_2.fastq.gz SRR10822356 SRR10822356_1.fastq.gz SRR10822356_2.fastq.gz SRR10822357 SRR10822357_1.fastq.gz SRR10822357_2.fastq.gz SRR10822358 SRR10822358_1.fastq.gz SRR10822358_2.fastq.gz SRR10822359 SRR10822359_1.fastq.gz SRR10822359_2.fastq.gz SRR10822360 SRR10822360_1.fastq.gz SRR10822360_2.fastq.gz解析にはgzip圧縮された状態のファイルも使用可能ですので、この後は「fastq.gz」ファイルを用いて解析を行っていきます。
まとめ
- Anacondaのcondaコマンドで、SRA Toolkitをインストール
- SRA Toolkitに含まれるprefetchコマンドで、SRAファイルをダウンロード
- SRA Toolkitに含まれるfasterq-dumpで、SRAファイルをFASTQファイルに変換
- pigzコマンドで、FASTQファイルをgzip圧縮
※ 本記事で使用したコマンドの使い方まとめ
# カレントディレクトリのファイルの一覧
% ls
# 新しいファイルを作成
% touch ファイル名
# ファイルを開く
% open ファイル名
# テキストファイルの中身を確認
% cat ファイル名
# 指定したSRRデータをダウンロード
% prefetch SRR番号
# ’ --option-file ’オプションを使用して、テキストファイルに記述されたSRRデータを全てダウンロード
% prefetch --option-file テキストファイル名
# Anaconda.orgから(https://anaconda.org/)からパッケージ名を検索すると、インストール用コマンドが出てくるので、そのコマンドをターミナルに打ち込む
% conda install パッケージ名
# Homebrewのインストール
% /bin/bash -c "$(curl -fsSL https://raw.githubusercontent.com/Homebrew/install/HEAD/install.sh)”
# Homebrew公式HP(https://brew.sh/index_ja)でパッケージ名を検索すると、インストール用コマンドが出てくるので、そのコマンドをターミナルに打ち込む
% brew install パッケージ名
# SRA形式をFASTQ形式に変換
% fasterq-dump SRR番号
# SRA形式をFASTQ形式に変換
% fasterq-dump SRR番号
# gzip圧縮
% pigz ファイル名