はじめに
前回はFATSQファイルのダウンロードまで行いました。今回は下記の記事の続きとなります。

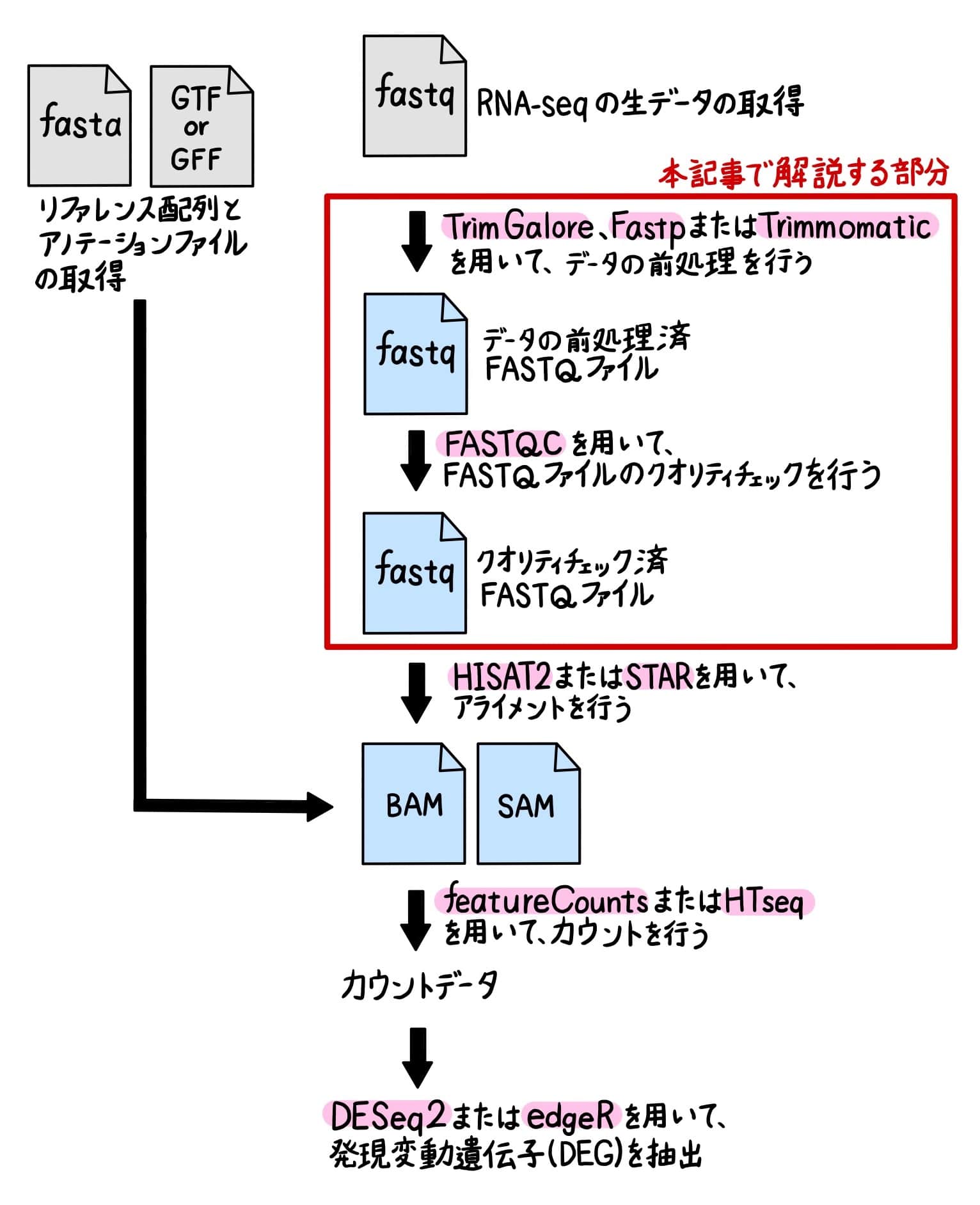
次世代シークエンサーまたは公共データベースから取得したFASTQファイルは、アダプター配列や低品質なリードを含んでいます。したがって、FASTQファイルを取得した後は、Trim Galore、Fastp、Trimmomaticなどのツールを用いて、不要な配列の除去やクオリティフィルタリングなどの前処理を行う必要があります。
今回はFASTQファイルの前処理・クオリティチェックについて、実際の論文データを用いて詳しく解説していきます(図1)。
実際、近年は次世代シークエンサーの発達に伴い、QCチェックで落ちることはほとんどありません。
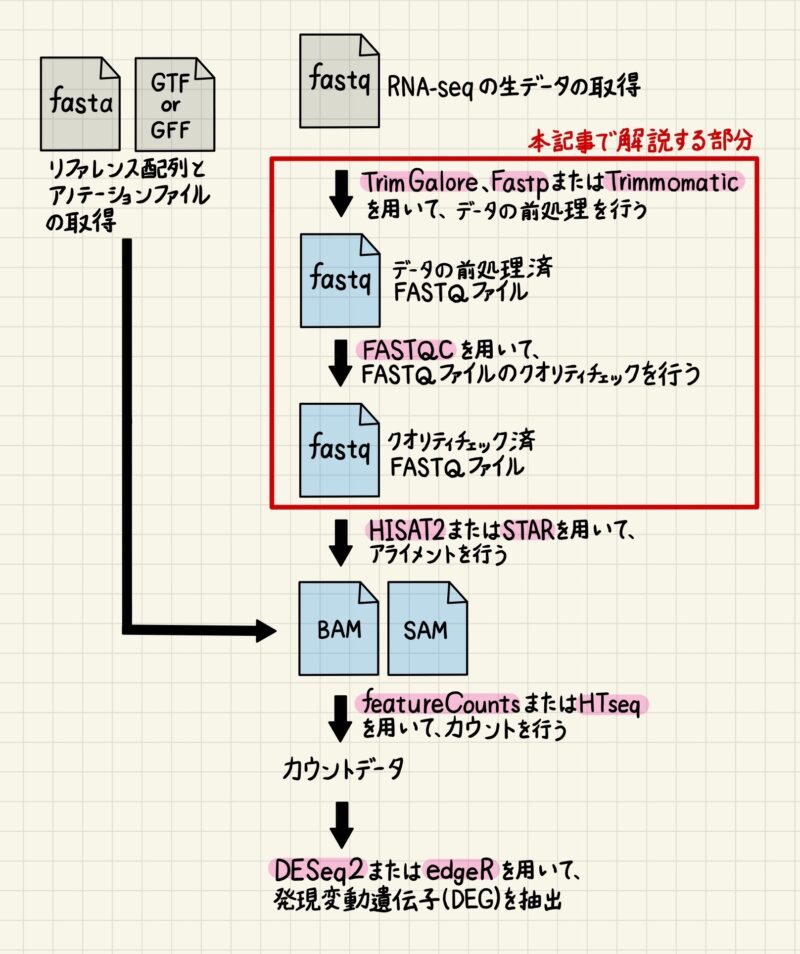
※ RNA-seq解析(ドライ解析)全体の流れは下記の記事で詳しく解説しています。
- PC: MacBook Pro 14インチ、2021
- チップ: Apple M1 Pro
- メモリ: 32 GB
- OS: macOS Ventura 13.4.1
- ストレージ: 512 GB
- ターミナル: zsh
fastpインストール
これまでは、アダプター配列の除去などの前処理には「Trimmomatic」、FASTQファイルのクリティチェックには「FASTQC、というように複数のツールを組み合わせる必要がありました。
fastpというツールはFASTQCやTrimmomaticなどの機能を備えており、1つのツールでFASTQファイルの前処理・クオリティチェックを行うことができます。また、fastpはそれぞれを単独で使用した場合よりもはるかに高速に動作します。
ですので、今回はfastpを用いたFASTQファイルの前処理・クオリティチェックのやり方を解説します。
まずはcondaコマンドを用いて、fastpをダウンロードします。
# Anacondaに含まれるcondaコマンドで、 fastpをダウンロード
conda install -c bioconda fastpcondaコマンドでエラーが出る場合
condaコマンドで「PackagesNotFoundError」というエラーが出る場合は、Homebrewのbrewコマンドを用いてfastpをインストールします。
# brewコマンドでfastpをインストール
% brew install fastpbrewコマンドでエラーが出る場合
従来、Apple社のMacBookには、主にIntel製の「x86_64」アーキテクチャを採用したプロセッサが搭載されていました。2020年以降のMacBookには、「x86_64」代わって新たに「arm64」というARMアーキテクチャを採用したプロセッサ(M1、M2)が搭載されています。
しかしながら、現時点ではARMアーキテクチャに対応していないアプリケーションも多く存在します。したがって、 M1搭載MacBookユーザーは、「arm64」未対応のアプリケーションを使用する場合はIntelアーキテクチャ(x86_64アーキテクチャ)環境を構築する必要があります。
現在は「x86_64」から「arm64」への移行段階であり、将来的には「arm64」のアプリケーションが主流になっていくと思われます。そのため、「arm64」対応アプリケーションは「arm64」環境で使用し、「arm64」に未対応のアプリケーションは「x86_64」で使用するという方針が良いでしょう。
2023年7月現在、fastpも「arm64」環境ではHomebrewでインストールできないようです。実際に brewコマンドを用いてfastpをインストールしようとすると、下記のようなエラーが出ます。エラー内容を見ると、「x86_64アーキテクチャ」が必要だと書いてあります。
% brew install fastp
==> Downloading https://formulae.brew.sh/api/formula.jws.json
==> Downloading https://formulae.brew.sh/api/cask.jws.json
fastp: The x86_64 architecture is required for this software.
isa-l: The x86_64 architecture is required for this software.
Error: fastp: Unsatisfied requirements failed this build.このエラーに対処するために、「x86_64」環境で動作するHomebrewをインストールします。まず、name -mコマンドで現在のアーキテクチャを確認します。デフォルトでは「arm64」となっているはずです。 arch -x86_64 zshコマンドで「x86_64」に切り替えます。そして、「x86_64」環境で、Homebrewをインストールします。これで、brewコマンドでfastpをインストールできるようになります。
しかし、このままでは「arm64」と「x86_64」環境用の両方のHomebrewが混在している状態であり、今後のアプリケーション管理がかなり複雑化してしまいます。そこで「arm64」と「x86_64」を適切に使い分ける”仕掛け”が必要です。
# unameコマンドで、現在のアーキテクチャを表示
% uname -m
arm64
# 「arm64」から「x86_64」への切り替え
% arch -x86_64 zsh
# unameコマンドで、「x86_64」へ切り替わっていることを確認
% uname -m
x86_64
#「x86_64」環境で動作するHomebrewをインストール
/bin/bash -c "$(curl -fsSL https://raw.githubusercontent.com/Homebrew/install/HEAD/install.sh)"※ Homebrewインストール用のコマンド「/bin/bash -c “$(curl -fsSL https://raw.githubusercontent.com/Homebrew/install/HEAD/install.sh)”」は今後変更される可能性もあるので、 インストールの際はHomebrewの公式ホームページ(https://brew.sh/index_ja)を確認してください。
「arm64」と「x86_64」を使い分ける方法いくつかありますが、下記の記事がシンプルで環境汚染もほとんどなく、非常にわかりやすかったので、同じ方法を採用します。詳しくは引用元の記事をご覧ください。

簡単に説明しますと、openコマンドで、ホームディレクトリ直下にある zprofileおよびzshrcファイルを開き下記のコードを入力します。すると、ターミナルログイン時に「arm」または「x86_64」と入力することで、arm64環境とx86_64環境を簡単に使い分けることができます。
# openコマンドで、zprofileおよびzshrcファイルを開く
% open ~/.zprofile
% open ~/.zshrczprofileファイルには下記のコードを入力します。
ARCH=$(uname -m)
if [[ $ARCH == arm64 ]]; then
echo "Current Architecture: $ARCH"
eval $(/opt/homebrew/bin/brew shellenv)
elif [[ $ARCH == x86_64 ]]; then
echo "Current Architecture: $ARCH"
eval $(/usr/local/bin/brew shellenv)
fizshrcファイルには下記のコードを入力します。
alias arm="exec arch -arch arm64 /bin/zsh --login"
alias x64="exec arch -arch x86_64 /bin/zsh --login"FASTファイルの前処理、クオリティチェック
現在、ほとんどの実験はペアエンドでシークエンスを行います。
ペアエンドシークエンスの場合、次のようにコマンドを入力してFASTファイルの前処理、クオリティチェックを行います。リード1のFASTQファイルを「-i」の後に、リード2のFASTQファイルを「-I」の後に書きます。前処理後のリード1のFASTQファイルを「o」の後に、リード2のFASTQファイルを「-O」の後に書きます。今回は前処理後のファイル名をそれぞれ「SRR10822355_filt_1.fastq.gz」「SRR10822355_filt_2.fastq.gz」としました。
前処理の結果はターミナルにも出力されますが、html形式のレポートも出力されます。このようにして、他のリードに対しても同様に名処理およびクオリティチェックをしていきます。
# fastpコマンドを用いて、FASTQファイルを前処理を行う
% fastp -i SRR10822355_1.fastq.gz -I SRR10822355_2.fastq.gz -o SRR10822355_filt_1.fastq.gz -O SRR10822355_filt_2.fastq.gzレポートの解釈
fastpコマンドでデータの前処理を行うと「fastp.html」という名前のファイルが出力されます。
それではopenコマンドで「fastp.html」を開き、前処理結果を確認していきます。
# openコマンドを用いて、前処理結果ファイルを開く
% open fastp.htmlSummary
レポートを開くと初めにSummaryを見ることができます。
Summaryを見ると150bpのペアエンドシークエンスが行われ、フィルタリング後はアダプターなどの不要な配列が除去されて、リード長が平均149bpとなっていることがわかります。
Q20 bases、Q30 basesはクオリティスコア(Qスコア)と呼ばれ、読み取った塩基の精度を示しています。Q10で90%、Q20で99%、Q30で99.9%の精度を表します。一般的に、次世代シークエンスにはQ30以上のスコアが求められるのですが、フィルタリング後のQスコアが94%と、ほとんどQ30以上なので問題なさそうです。
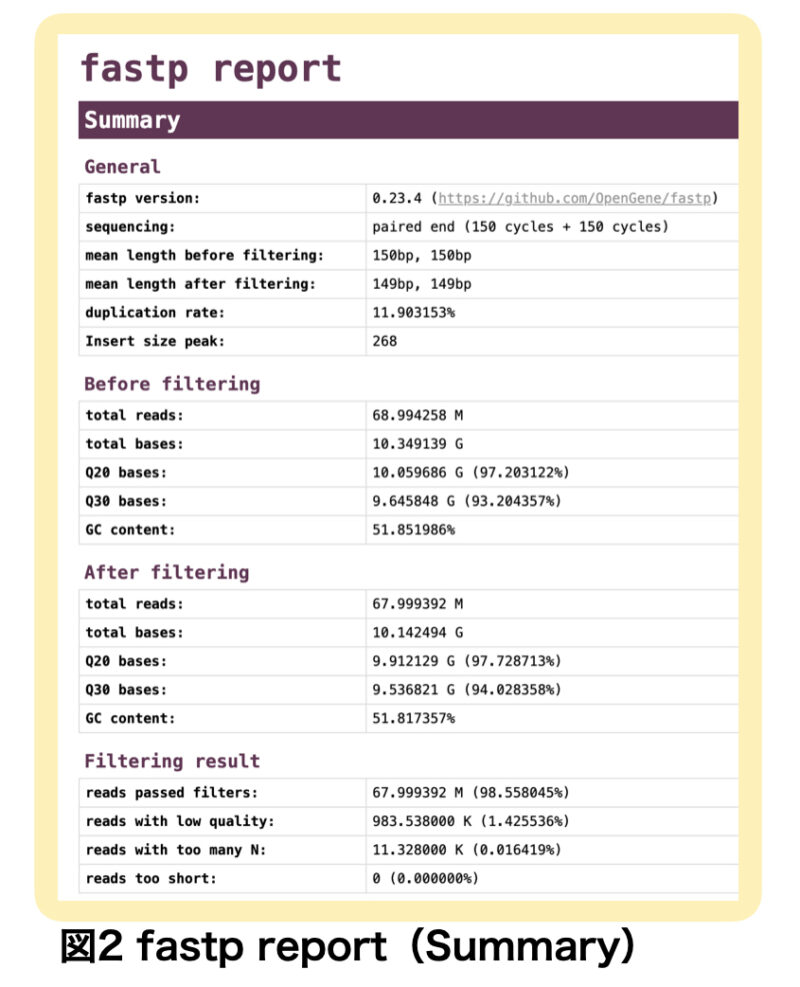
Duplication rate(重複率)は複数回シークエンスされたフラグメントの割合を示しています。
重複率は増幅エラーなどの技術的な要因で高くなる場合があります。重複率が高いとシークエンスデータの情報量が減少してしまうので、高いDuplication rateは好ましくありません。しかし、RNA-seqでは同じ遺伝子領域からの発現が高ければ重複率は上昇するわけで、重複率が生物学的な意味を持っている可能性があります。
RNA-seq解析においては、重複したインサートを解析から除去すると、精度や正確さは向上せず、むしろ発現変動遺伝子(DEG)の検出力と偽発見率 (FDR) を悪化させる可能性があるということが指摘されており、一般的に重複は除去しない方が良いでしょう。
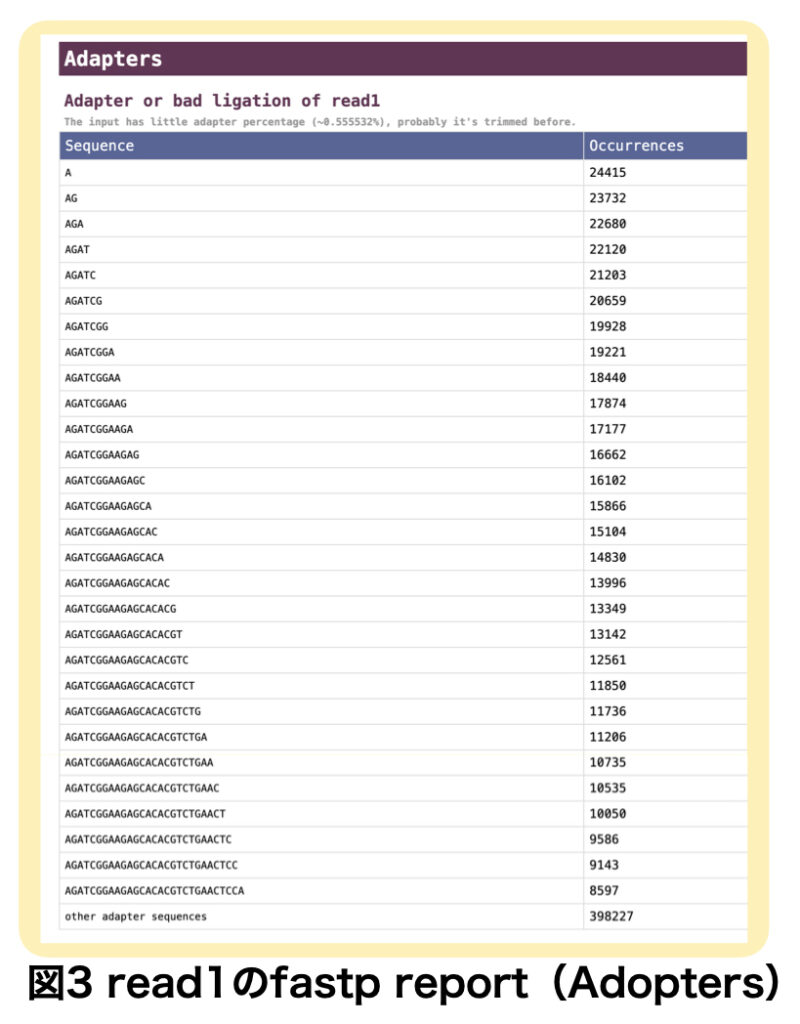
Adapters
fastpはアダプター配列を自動的に検出し、トリミングを行います。シークエンスデータの~0.555532%がアダプター配列としてトリミングされたことがわかります。
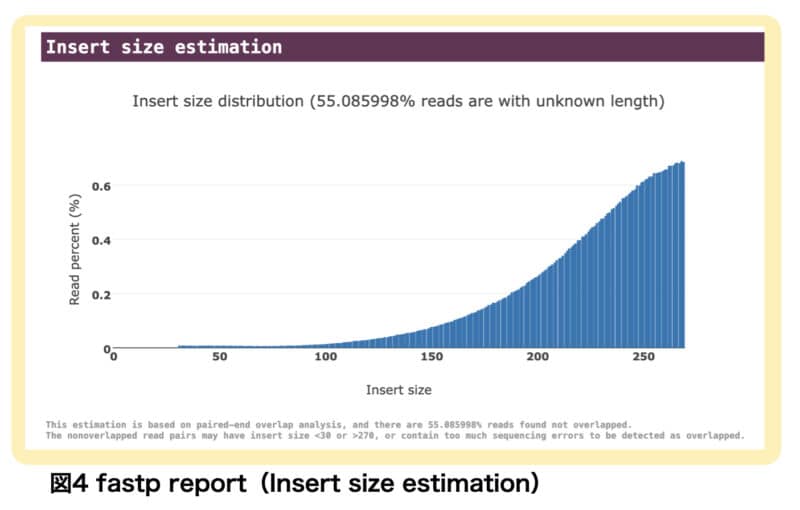
Insert size estimation
インサートのサイズを表しています。今回使用した論文では、インサートのサイズが大きく図が途中で途切れていますが、インサートの推定サイズは300bp程度だということがわかります。
Before filtering、After filtering
quality
「Before filtering:read1:quality」には、縦軸に品質スコア、横軸にリード中の塩基の位置を示したグラフが描かれています。蛍光の減衰などにより、端の方に進むほど読み取り精度は落ちていきます。また、PCRステップでランダムプライマーを用いていることが原因で、リードの最初の数塩基も読み取り精度は低くなります。
縦軸に注目すると、リード末端でもQスコアは38程度であり、Beforeでも十分に読み取り精度が高いことがわかります。「After filtering:read1:quality」を見ると、少しQスコアが改善していることが分かります。
塩基の読み取り精度は100%ではないので、同じ領域を繰り返しシークエンスして精度を高めるということが行われます。このシークエンスの回数を被覆率(coverage)といい、一般的に30回被覆されます。
Base contents
「Before filtering:read1:Base contents」は、読み取られた塩基の割合を示しています。
リードの開始部分はライブラリ調製ステップで起こる秘術的な問題によりブレていますが、ほとんどの場合下流の解析に悪影響を与えるものではなく、気にする必要はないようです。
一般的に、シークエンス反応が進むにつれ、 TおよびCが誤ってGとして検出されることが多くなります(polyG tail issue)。したがって、リード末端に進むにつれて、Gの曲線は右肩上がりになることが多いです。今回はBeforeであっても、リード末端まで読み取られた塩基の割合は一定であり、良好なデータであると言えます。
KMER counting
リード中のk個の連続した塩基のことをK-merというのですが、「Before filtering:read:KMER counting」は様々なK-merの出現頻度を濃淡で表現しています。色が濃いほど、そのK-mer配列の出現頻度が多いということを意味しています。図では、k=5のk-merが表示されています。K-merの出現頻度の偏りは、サンプリング、断片化、PCR、コンタミなどが原因である可能性があります。
重複率と同様、遺伝子発現が高ければ特定のK-merの出現度も高くなります。
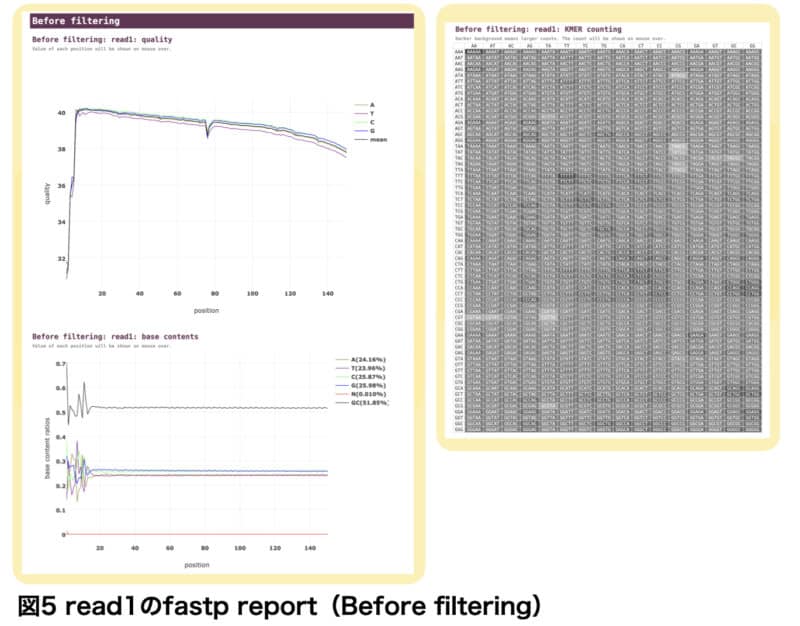
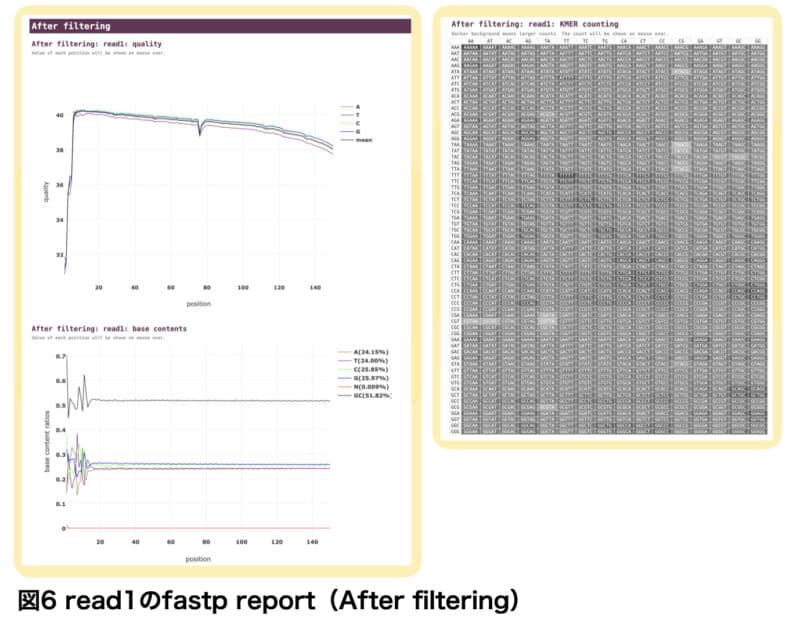
※ 図5、6にはReas1のみを図示しています。
まとめ
- Anacondaのcondaコマンドで、fastoをインストール
- fastpコマンドでFASTQファイルの前処理を行う
- openコマンドで、前処理結果の確認・クオリティチェックを行う
# Anaconda.orgから(https://anaconda.org/)からパッケージ名を検索すると、インストール用コマンドが出てくるので、そのコマンドをターミナルに打ち込む
% conda install パッケージ名
# Homebrew公式HP(https://brew.sh/index_ja)でパッケージ名を検索すると、インストール用コマンドが出てくるので、そのコマンドをターミナルに打ち込む
% brew install パッケージ名
# unameコマンドで、現在のハードウェア(アーキテクチャ)を表示(-mはコンピュータのハードウェアを表示するオプション)
% uname -m
# アーキテクチャを「x86_64」へ切り替え
% arch -x86_64 zsh
# openコマンドを用いて、ファイルを開く
% open ファイル名など- Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018 Sep 1;34(17):i884-i890. doi: 10.1093/bioinformatics/bty560. PMID: 30423086; PMCID: PMC6129281.
- ‘t Hoen PA, Friedländer MR, Almlöf J, et al. Reproducibility of high-throughput mRNA and small RNA sequencing across laboratories. Nat Biotechnol. 2013;31(11):1015-1022. doi:10.1038/nbt.2702